First Total Synthesis of Darwinolide (M. Christmann, 2018)
The diterpenoid darwinolide (1) is a promising candidate for antibiotic research due to his antibiofilm properties. The natural product was isolated in 2016 from Dendrilla Membranosa. The structure features an unusual tricyclic system attached to a cyclohexyl moiety. Prof. M. Christmann et al. from the Freie Universität Berlin now published the first asymmetric total synthesis in Angewandte Chemie.
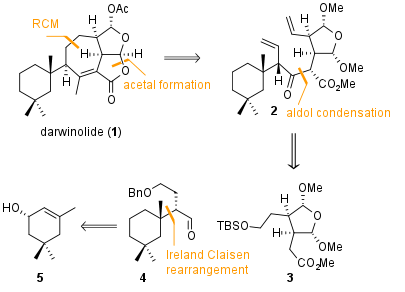 Figure 1: Retrosynthetic analysis of darwinolide (1).
Figure 1: Retrosynthetic analysis of darwinolide (1).

metathesis.metathesis as shown in Figure 1. Also, one of the two five membered cyclic lactones should be closed in a late stage transformation. The precursor 2 can be coupled by aldol condensation from the two fragments 3 and 4. They are readily available in a few steps from literature known compounds.
Synthesis of Fragment 4
The key step for the synthesis of fragment 4 isis, as shown in Scheme 1, an Ireland Claisen rearrangement from compound 6. This fragment itself is synthesized by Steglich esterification from literature known alcohol 5. Substrate 6 was then enolized to intermediate 7 which was then subjected to rearrangement conditions to yield fragment 8 in 71% yield. Although the stereocenter in alpha position to the acid had to be corrected later in the synthesis, this rearrangement could provide the precursor for fragment 4, which was available after reduction and oxidation under Stahl's conditions in 70% over 2 steps.

 Scheme 1: Synthesis of fragment 4 by Claisen rearrangement.
Scheme 1: Synthesis of fragment 4 by Claisen rearrangement.
Synthesis of Fragment 3
Commercially available acid anhydride 9 was reduced to the diol followed by re-oxidation under Swern conditions and acetalization in 61% yield over 2 steps.steps (see Scheme 2). Sharpless dihydroxylation gave 11 in 86% yield and 7:1 diastereomeric ratio. Note that compound 11 is achiral and only the desymmetrization due to the proline derived catalyst led to chiral fragment 12 in 83% yield and 96% ee. Oxidation and deprotection gave 13 which was then subjected to Pb(OAc)4 which led to C-C bond cleavage. The substrate 14 could be synthesized in 61% yield over 3 steps. Final reduction of the aldehyde and TBS protection delivered the desired fragment 3.
 Scheme 2: Synthesis of fragment 3 starting with symmetrical anhydride 9.
Scheme 2: Synthesis of fragment 3 starting with symmetrical anhydride 9.


With both fragments in hand, the authors coupled the fragments by aldol addition promoted by NaHMDS in 42% yield.yield as shown in Scheme 3. Note that fragment 15 was synthesized as a diastereomeric mixture which was used as a mixture for the synthesis in the next 7 (!) steps. After oxidation (88%) both protecting groups were removed under reductive (OBn) and acidic (OTBS) conditions. The primary alcohols of 16 were then subjected to a three-step olefination protocol which led to the RCM precursor 2 in 72% yield.
 Scheme 3: Completion of total synthesis of darwinolide (1).
Scheme 3: Completion of total synthesis of darwinolide (1).
Fragment 2 was then cyclized by metathesis using Umicore's Hoveyda Grubbs Catalyst M710 (CAS: 1025728-56-6) in an impressive 86% yield. Now epimerization with C2CO3 took place which set the final stereochemistry. Olefin 17 was then reduced and enolate 18 was synthesized and directly subjected to cross-coupling conditions to install the required methyl group. Due to the Lewis acidic conditions, the final tricyclic ring system was build up spontaneously in 40% overall yield. Finally, trans acetalization led to natural product darwinolide (1).
In the end, the authors successfully synthesized darwinolide (1) in 21 steps with a total yield of 1.4%. Although only 0.8 mg could be achieved, this amount was enough to support the identity of the substrate.
Published in Angewandte Chemie Int. Ed. 2018 by T. Siemon, S. Steinhauer, M. Christmann Angew. Chem. Int. Ed. 2018, 58, 1120-1122. doi: 10.1002/anie.201813142