Synthesis Strategies for Entecavir
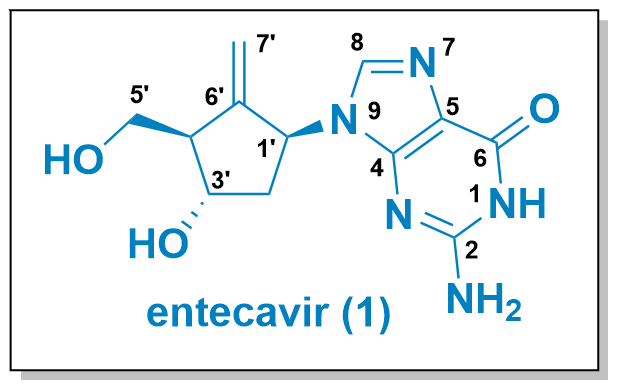
Entecavir (1, see Figure 1) is a carbocyclic nucleoside analogue designed to mimic the structural characteristics of guanosine. Unlike the ribose-based natural guanosine, in entecavir the ring oxygen is replaced with a carbon atom, resulting in a more metabolically stable analogue. However, this substitution often introduces an unfavourable structural conformation. To address this, an exocyclic double bond locks the carbocycle into a ribose-like structure, thereby achieving a favourable conformation. First described by Bristol-Myers Squibb in 1988, entecavir is approved as a potent antiviral medication for the treatment of Hepatitis B virus infection.
Figure 1 illustrates various synthetic routes available for the synthesis of entecavir. Unlike ribose-type nucleoside analogues that can be synthesized in a few steps from natural precursors, constructing the carbocyclic core of entecavir is a more intricate process. In an early patent by Bristol-Myers Squibb in 1988, sodium cyclopentadienyl was functionalized to obtain the desired compound 1.[1,2] Kawal reported an alternative synthesis route using substituted cycloketone 3, which can be derived from ribose.[3] Liu and later Valasco also employed acyclic precursors, such as 4 and 5, respectively.[4,5]
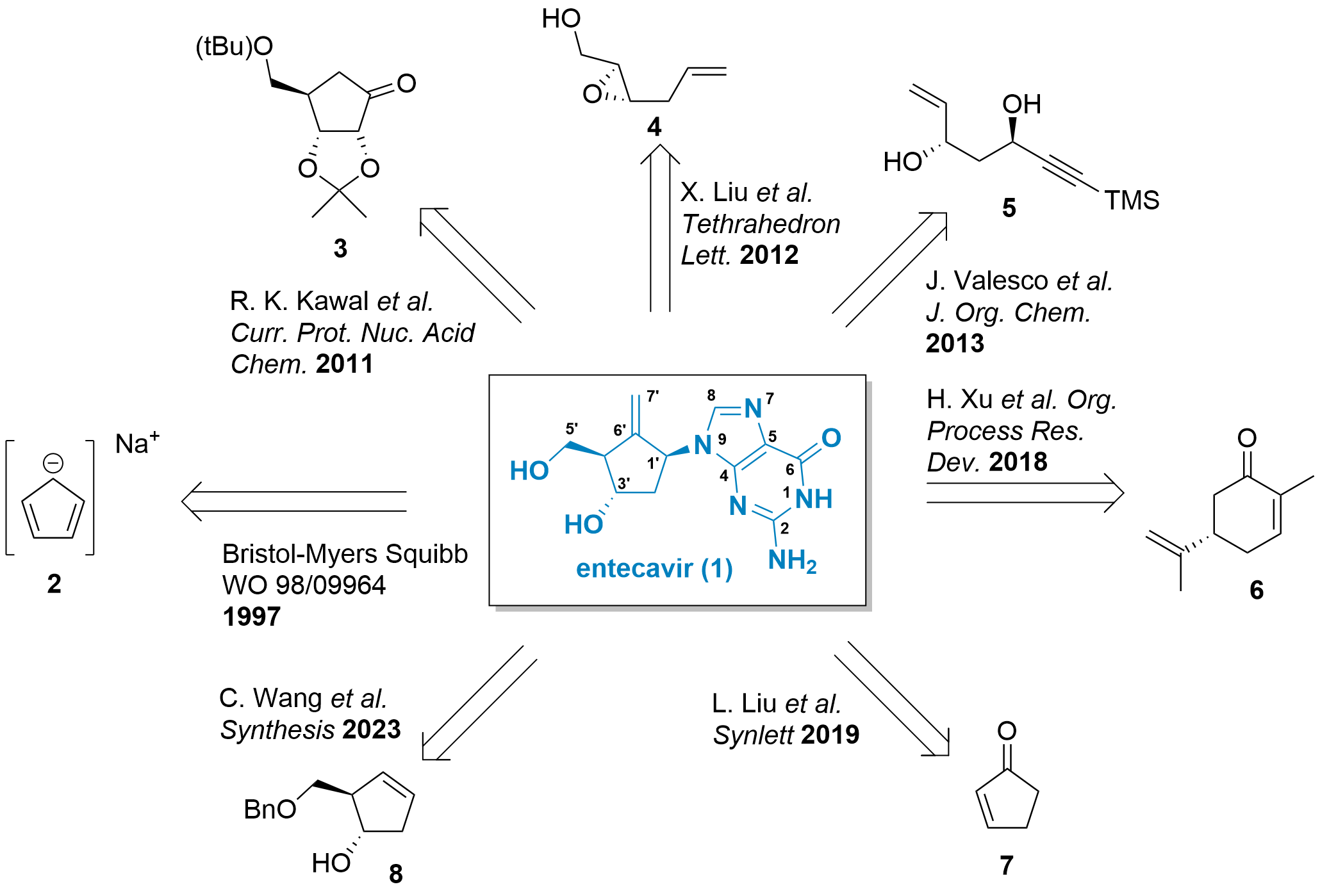
Figure 1: Structure of entecavir and synthetic strategies for its synthesis.
In a different approach, Xu described a kilogram-scale synthesis of entecavir starting from carvone 6 in 2018.[6] Another strategy was reported by Liu, utilizing optically inactive cyclopentenone 7.[7] Lastly, Wang presented a recent development in 2023, focusing on the synthesis of entecavir analogues starting from intermediate 8, derived from the Bristol-Myers Squibb synthesis.[8]
These diverse synthetic strategies highlight the ongoing efforts to optimize the synthesis of entecavir and explore alternative routes. Each approach offers its own advantages and challenges, contributing to the overall understanding and advancement of the synthetic methodologies employed in the production of entecavir and its analogues.
Employing Sodium Cyclopentadienyl as the Core Structure
Bristol-Myers Squibb initially disclosed the synthetic route of entecavir in a patent in 1988.[1] A slightly improved version of the route was also published in the journal Biorg. Med. Chem. Lett. in 1997, as shown in Scheme 1[2] First, sodium cyclopentadienyl (2) is functionalized by coupling with BnOCH2Cl and further reduction with pinene derived asymmetric borane yielding sugar-type analogue 8 in 75% yield. Then, diastereoselective epoxide formation followed by benzylation leads to the formation of compound 9, with an 83% yield. The protected guanine 10 is activated with lithium hydride, and the subsequent epoxide opening yields compound 11. The free amine of compound 11 is protected with MMT for the subsequent oxidation with DMP to yield compound 12. The authors noted that the cyclopentanone moiety is prone to beta elimination, and only oxidation with the Dess-Martin periodinane provides satisfactory yields. Finally, Nysted methylenation of compound 12 produces compound 13, with a 75% yield. To obtain entecavir 1, the authentic guanine fragment is obtained through acidic treatment, and the Lewis acidic benzyl cleavage liberates the 1,3-diol moiety.
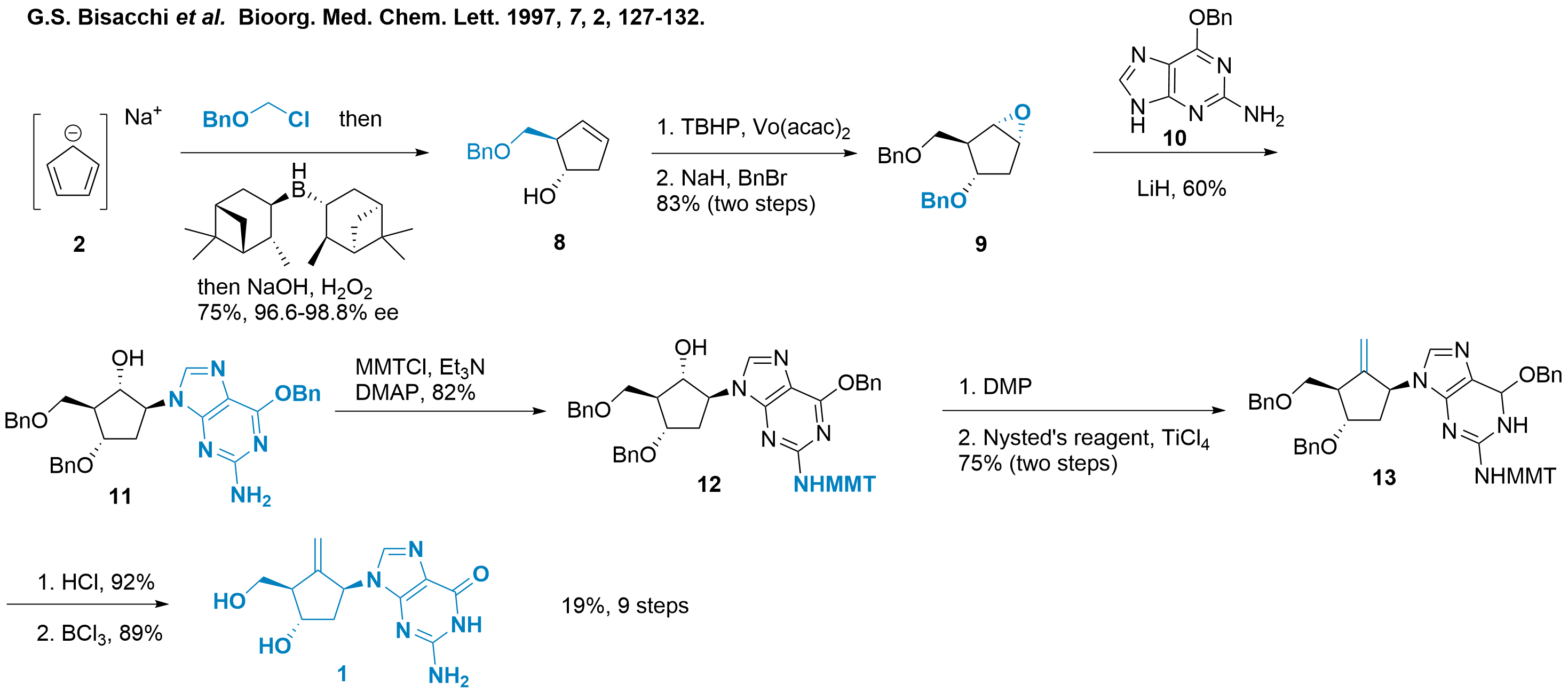
Scheme 1: Synthesis of entecavir (1) by functionalizing cyclopentadienyl (2).
In summary, this synthetic route delivers entecavir 1 in only 9 steps with high efficiency (19%). Despite the need for protection and deprotection steps and the use of several metal catalysts, this synthetic route is still considered elegant and capable of producing multigram amounts of the nucleoside analogue.
Functionalization of Cyclopentanone
Chung K. Chu and co-workers from the University of Georgia (USA) followed up with a synthesis starting with (D)-ribose in 2011.[3] This approach allows the usage of cheap and available starting material. Furthermore, the stereoconfiguration is already set, therefore no enantioselective reagent is necessary. However, since entecavir is a DNA analogue, deoxygenation is required. In addition, the initial functionalisation of ribose to obtain compound 3 involves a relatively complex process consisting of eight steps.[9]
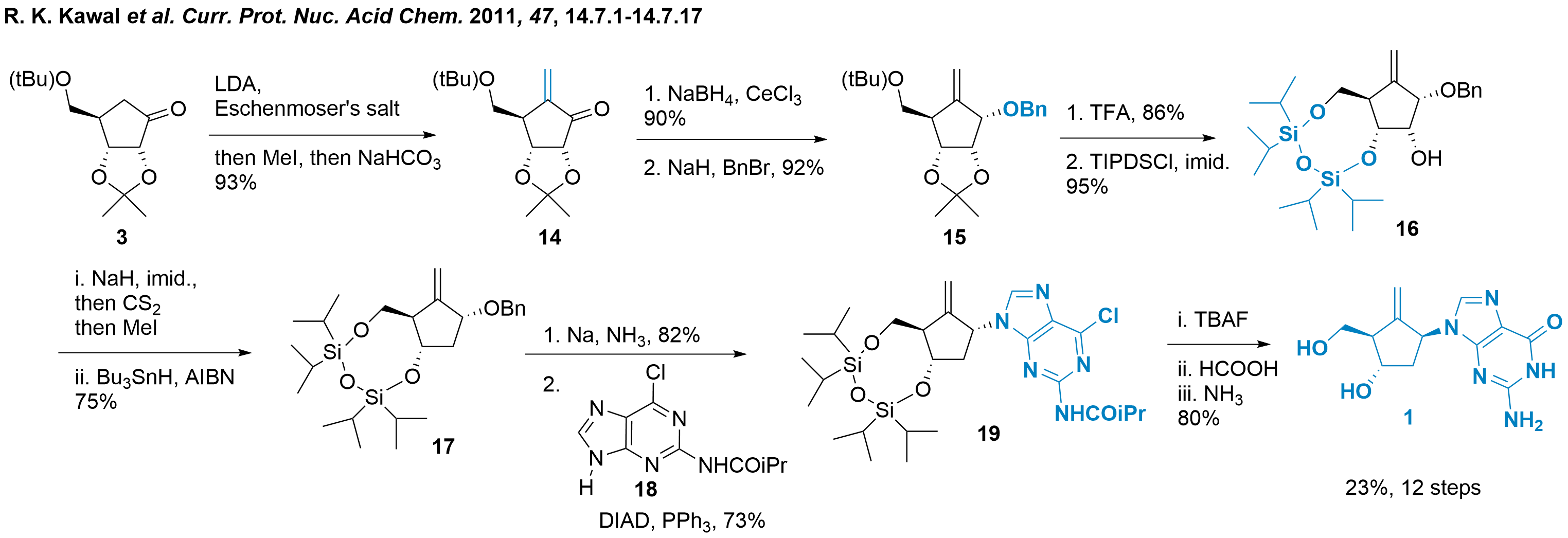
Scheme 2: Synthesis of entecavir from ribose by Chu and co-workers.
Once compound 3 is obtained, the formation of the exocyclic methylene group is achieved through enolate formation and treatment with Eschenmoser's salt as shown in Scheme 2. The formation of the ammonium salt subsequently triggers an elimination, resulting in compound 14 with an overall yield of 93%. A diastereoselective reduction is performed to generate the free alcohol at the C1' position, which is then protected as a benzylate in compound 15. The tert-butyl and acetonide protecting groups are removed through acid deprotection using TFA, leading to the formation of triole. By employing a literature-known protection method, compound 16 is obtained, providing selective access to the 2'-alcohol. To achieve deoxygenation and obtain the DNA-like fragment 17, a commonly used two-step radical procedure is employed, resulting in a yield of 75%. Note, that this synthetic strategy requires a total of 15 steps from ribose to construct the core structure 17 of the carbasugar moiety. With this compound in hand, reductive deprotection and Mitsunobu coupling reactions are carried out, leading to the formation of protected entecavir compound 19. Finally, fluoride-mediated deprotection and guanine modification ultimately yielded entecavir in 12 steps from compound 3 (or 20 steps from ribose).
Epoxide Opening and Ring Closing Metathesis
In 2012, Ping Xie and colleagues from the Institute of Materia Medica (Peking, China) reported a concise synthesis of entecavir (1) from acyclic precursor 4 as shown in Scheme 3.[4] The precursor 4 itself is literature known and available in two steps by coupling[10] and Sharpless epoxidation.[11] Then, organometallic epoxide opening led to the diol 20 in a diastereoselective manner. The primary alcohol was TBS protected and ring-closing metathesis gave 21 in high yields. The C3'-OH-directed epoxidation and TBS protection gave 22. For installment of the exocyclic double bond, epoxide isomerisation was triggered under Lewis acidic conditions yielding in core structure 23 in again excellent yield. Then, coupling with guanine 24 by using Mitsunobu conditions furnished 25 which can be transformed into entecavir (1) by acidic treatment.
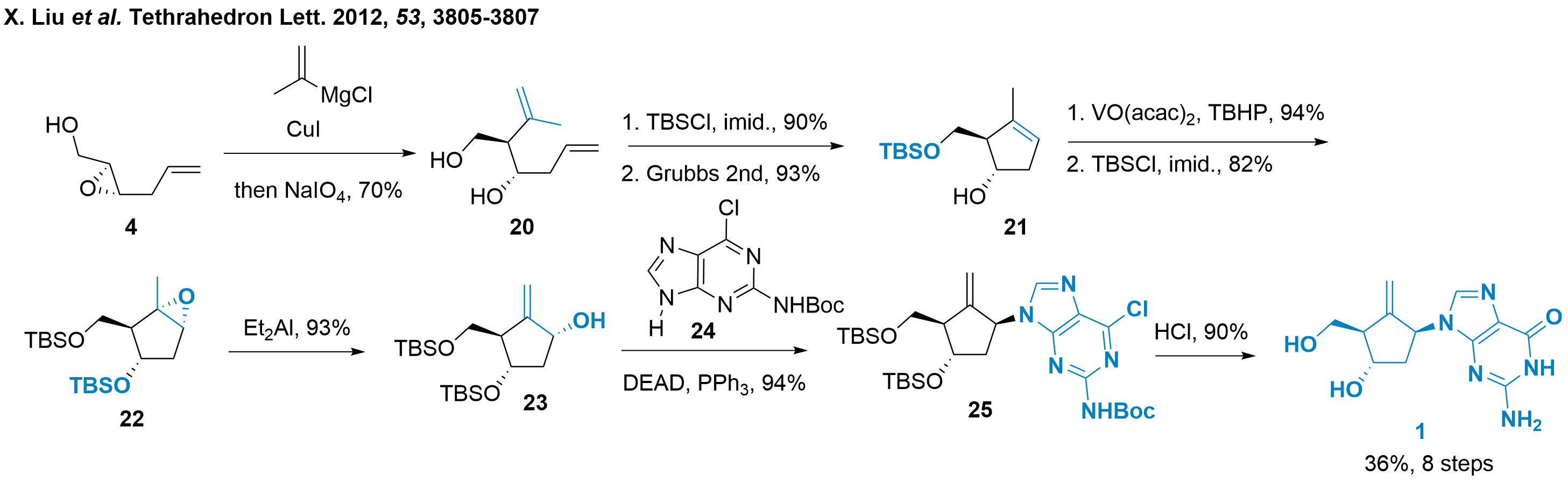
Scheme 3: Xie's synthesis for entecavir (1) delivers the analogue in eight steps.
In conclusion, Xie and coworkers finished the synthesis in only eight steps from commercially available starting material. The synthesis is not only short but also uses robust reactions with high yields. The synthesis relied on the initial absolute configuration of 4 to establish the stereochemistry of other stereocenters in a diastereoselective manner.
Radical Cycloaddition
An alternative synthesis involving an acyclic precursor was reported by Yolanda Gasanz and colleagues from the Universitat de Barcelona (Spain). The key step in this synthesis is a radical cycloaddition reaction, which produces the desired cyclopentane 30, as depicted in Scheme 4.[5] Initially, an achiral butynone 26 was selected as the starting material. An asymmetric Aldol addition using pinene-derived DIPCl and acrolein was employed to generate the Aldol product in situ. This product was subsequently directly reduced, and the resulting boronate was oxidatively cleaved to form 1,3-syn diol 5. While this step established the desired stereoconfiguration, it exhibited moderate selectivity (dr 9:1, ee 60%). The enantiopurity of diol 5 was enhanced through recrystallization. Next, the less hindered alcohol was protected with a TBS group, followed by TMS alkyne cleavage to yield compound 27. Then epoxidation and acetylation finally gave the cyclisation precursor 28. The cyclisation was initiated by epoxide opening and radical formation, leading to the formation of compound 29, which upon cyclisation and radical quenching, yielded the desired product, cyclopentane 30. After several optimization attempts involving the use of an iridium catalyst, manganese, and collidine/TMSCl salts as additives, compound 30 was obtained with a 58% yield.

Scheme 4: Cycloaddition as the key step in the synthesis of entecavir (1) reported by Gasanz and co-workers.
With the core structure in hand, the primary alcohol was protected, and the C1'-OH group was deprotected to afford compound 31. A Mitsunobu reaction with guanine 32 was performed, resulting in the formation of compound 33, which was finally transformed into entecavir (1) using established procedures. This synthesis route is particularly noteworthy as it systematically explores the cycloaddition process with respect to the choice of protection groups and reagents. Consequently, it serves as a valuable reference for future synthetic projects.
Ring Contraction of Carvone
A completely different strategy was employed by Yehua Jin and colleagues from Launch-Pharma Technologies (Guangzhou, China) reported in 2018. In their strategy, they used carvone 6 as a readily available starting material as seen in Scheme 5. As carvone consists of cyclohexane, ring contraction is needed during this route.[6] The authors decided to achieve this through Favorskii rearrangement. To prepare this, carvone 6 was diastereo- and regioselective epoxied and acidic mediated giving the chlorohydrin which is readily transformed into the tosylate 34 in 60% yield in three steps. The exocyclic alkene was then epoxidized, opened, and protected towards Favorskii rearrangement precursor 35. The rearrangement was triggered to give the cyclopentene 36 after tosylate elimination. Reductive treatment liberated 5'-OH, which was followed by acidic deprotection and sodium periodate diol cleavage. The resulting ketone 37 was subjected to Bayer Villiger oxidation conditions and saponification yielded into the authentic 3'-OH. Then diastereoselective epoxidation gave core structure 38. From here, TBS protection and epoxide opening furnished 23 which transformation to 1 by Mitsunobu coupling with 24 and deprotection was already discussed in Scheme 3.
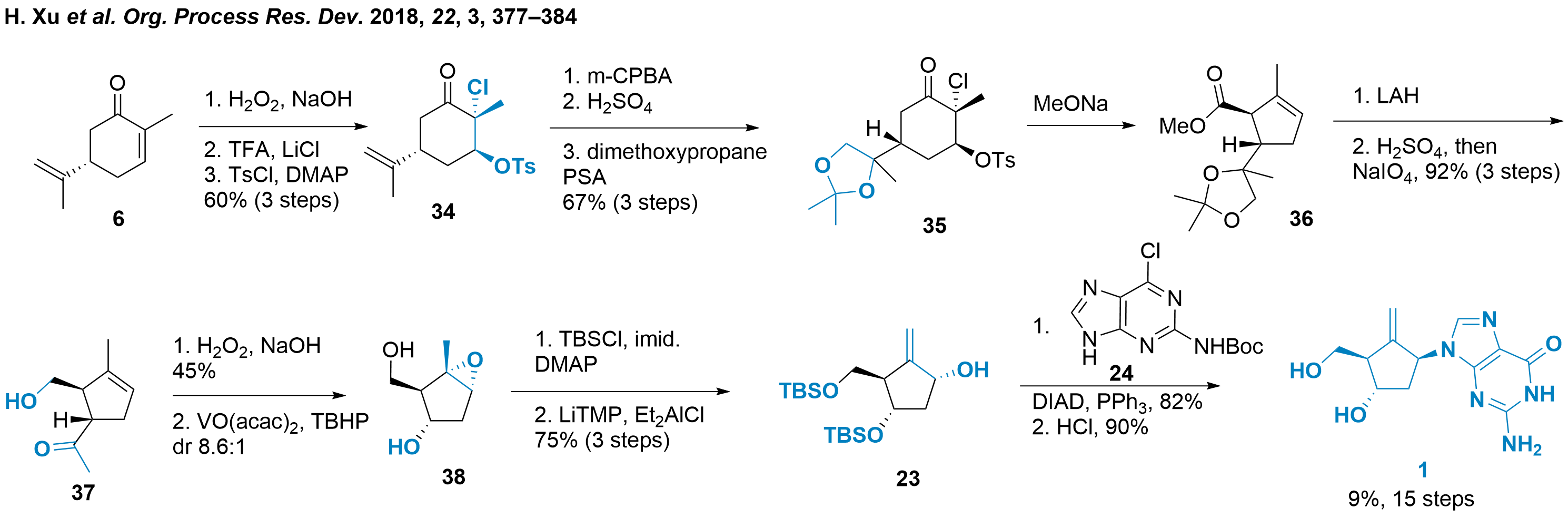
Scheme 5: Synthesis of entecavir by Favorskii rearrangement of carvone 6.
Even if this synthetic strategy is considered to be longer than previous routes, the ability to scale up was proven by the authors and 500 g of entecavir (1) could be synthesized in one batch.
Functionalising of Cyclopentenone
Another approach was chosen by Xiaoji Wang ( only as diastereomers as epimerization was observed. The authors described the elimination towards the cyclopentenone as a major side product in this step. After TBS protection of 3'-OH both isomers could be separated and methylenation gave 41 in 87% yield. The following C1' functionalization proved difficult in one step, as over-oxidation to the ketone and an isomeric mixture was observed. Therefore, a two-step protocol to the ketone and subsequent stereoselective reduction liberated 23 in moderate yield. As the synthesis of entecavir (1) from 23 is literature-known (see also Scheme 3 and 5), the authors concluded their studies here.
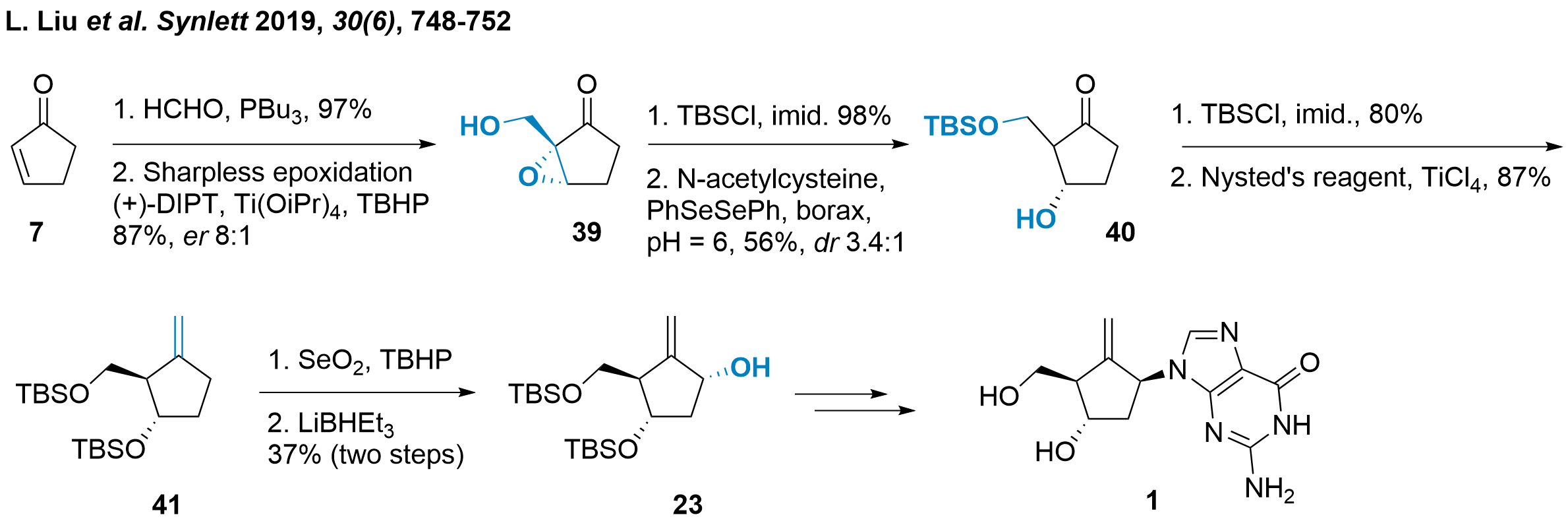
Scheme 6: Synthesis of entecavir by Xiaoji Wang and colleagues.
In conclusion, Wang's strategy to utilize unsubstituted cyclopentenone 7 is challenging, and several drawbacks have been observed during this synthetic route. The medium selectivity of the Sharpless epoxidation towards 39, the epimerization of compound 40 and the difficulties in oxidising compound 41 were major hurdles during this study. Aside from this, key intermediate 23 could be achieved in only 8 steps and it could be proved that a simple cyclopentenone could be used as a suitable starting material.
Synthesis of Entecavir 1'-Epimere
Finally, the synthetic strategy by Chao Wang and coworkers from Xihua University (Chengdu, China) will be discussed in Scheme 7.[8] It is important to see, that not entecavir (1) was targeted but epi-1'-entecavir 45, which should gain a better insight into the SAR of the compound. The authors started from common intermediate 8 (see also Scheme 1), which was diastereoselective epoxidised and protected to 9 similar to the synthesis of Bisacchi et al.[2] As the epimer is targeted, the epoxide can now be conveniently opened by the addition of tert-butanol. The remaining alcohol was oxidised using DMP and methenylation and tBu deprotection yielded 42 in overall good to excellent yields. Coupling with diprotected guanine 43 was achieved under Mitsunobu's conditions towards 44 and final deprotection and guanine functionalization gave epimer 45.
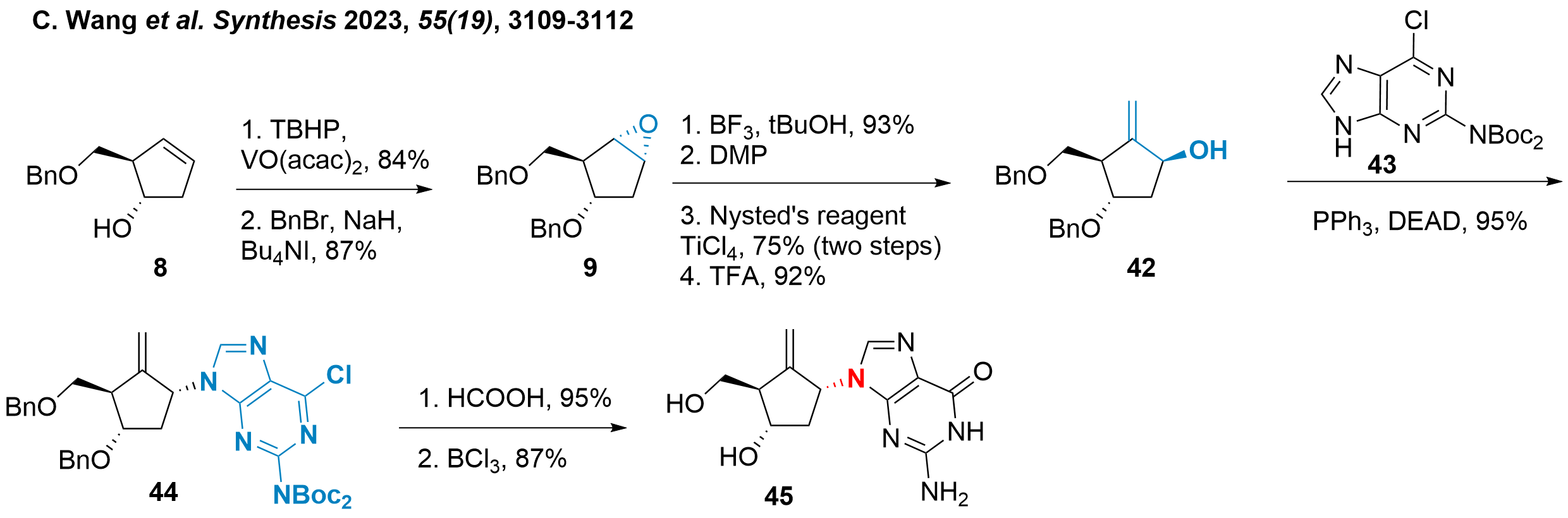
Scheme 7: Synthesis of epi-1'-entecavir (45).
Aside from 45 two other epimers were synthesized (not shown), however, their biological activity was not accessed. This, of course, would be of overall importance to assess the importance of these compounds and study.
Analytical Properties of Entecavir
The following 1H-NMR data is collected from literature:[3,12]
| 800 MHz, DMSO-d6[12] |
500 MHz, DMSO-d6[3] | |
| 2-NH2 |
6.41 (br s, 2H) | 6.56 (bs, 2 H) |
| 3-NH | 10.53 (s, 1H) | 10.69 (bs, 1H) |
| 8 |
7.66 (s, 1H) | 7.68 (s, 1H) |
| 1' | 5.36 (dddd, J=10.3, 7.6, 2.4, 2.4 Hz, 1H) | 5.35-5.39 (m, 1H) |
| 2' |
2.21 (ddd, J=12.6, 11.0, 4.6 Hz, 1H); 2.04 ppm (dddd, J=12.6, 7.7, 1.6, 1.6 Hz, 1H) |
2.20-2.26 (m, 1H, Ha) 2.03-2.07 (m, 1 H, Hb) |
| 3' |
4.23–4.22 (m, 1H) | 4.25 (bs, 1H) |
| 4' |
2.53–2.51 (m, 1H) | 2.52 (bs, 1H) |
| 5' |
3.55–3.51 (m, 2 H) | 3.52-3.55 (m, 2H) |
| 7' |
5.10 (t, J=2.3 Hz, 1H) 4.56 (t, J=2.3 Hz, 1H) |
5.11 (bs, 1H), 4.57 (bs, 1H) |
| 3'-OH |
4.87 (d, J=3.1 Hz, 1H) | 4.87-4.93 (m, 2H) |
| 5'-OH |
4.83 (t, J=5.4 Hz, 1H) | 4.87-4.93 (m, 2H) |
The 13C-NMR was reported as follows:
13C NMR (126 MHz, DMSO) δ 156.8, 153.4, 151.4, 151.3, 135.9, 116.2, 109.2, 70.4, 63.0, 55.1, 54.1, 39.2 ppm[6]
13C NMR (125 MHz, [D6]DMSO): δ=156.8, 153.4, 151.4, 151.3, 135.9, 116.2, 109.2, 70.3, 63.0, 55.1, 54.0, 39.8 ppm[12]
Literature
[1] G. S. Bisacchi, J. E. Sundeen 1997, Improved Process For Preparing The Antiviral Agent [1s-(1α, 3α, 4β)]-2-amino-1,9-dihydro-9-[4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-6H-purin-6-one, WO1998009964A, [link]
[2] G.S. Bisacchi, S.T. Chao, C. Bachard, J.P. Daris, S. Innaimo, G.A. Jacobs, O. Kocy, P. Lapointe, A. Martel, Z. Merchant, W.A. Slusarchyk, J.E. Sundeen, M.G. Young, R. Colonno, R. Zahler BMS-200475, a novel carbocyclic 2′-deoxyguanosine analog with potent and selective anti-hepatitis B virus activity in vitro. Bioorganic & Medicinal Chemistry Letters 1997, 7 (2), 127-132. 10.1016/S0960-894X(96)00594-X
[3] R. K. Rawal, U. S. Singh, S. Gadthula, C. K. Chu Synthesis of Entecavir and Its Novel Class of Analogs. Current Protocols in Nucleic Acid Chemistry 2011, 47: 14.7.1-14.7.17. 10.1002/0471142700.nc1407s47
[4] X. Liu, X. Jiao, Q. Wu, C. Tian, R. Li, P. Xie A novel and efficient synthesis of Entecavir. Tetrahedron Letters 2012, 53 (29), 3805-3807. 10.1016/j.tetlet.2012.05.058
[5] J. Velasco, X. Ariza, L. Badía, M. Bartra, R. Berenguer, J. Farràs, J. Gallardo, J. Garcia, Y. Gasanz Total Synthesis of Entecavir. The Journal of Organic Chemistry 2013, 78 (11), 5482-5491. 10.1021/jo400607v
[6] H. Xu, F. Wang, W. Xue, Y. Zheng, Q. Wang, F. G. Qiu, Y. Jin Total Synthesis of Entecavir: A Robust Route for Pilot Production. Organic Process Research & Development 2018, 22 (3), 377-384. 10.1021/acs.oprd.8b00007
[7] L. Liu, Y. Sun, J. Wang, W. Ou, X. Wang, S. Huang A New Formal Synthetic Route to Entecavir Synlett 2019, 30 (6), 748-752. 10.1055/s-0037-1612215
[8] C. Wang, D. Pei, L. Zhou, J. Sun Stereoselective Total Synthesis of Stereoisomers of Entecavir. Synthesis 2023, 55 (19), 3109-3112. 10.1055/a-2069-4552
[9] P. Li, G. Dräger, A. Kirschning A General Biomimetic Hetero-Diels–Alder Approach to the Core Skeletons of Xenovulene A and the Sterhirsutins A and B. Organic Letters 2019, 21 (4), 998-1001. 10.1021/acs.orglett.8b04003
[10] B. C. Ranu, A. Majee Indium-mediated regioselective Markovnikov allylation of unactivated terminal alkynes. Chemical Communications 1997, 1225-1226. 10.1039/A702241G
[11] M.-P. Heck, C. Baylon, S. P. Nolan, C. Mioskowski Triple Ring Closing Metathesis Reaction: Synthesis of Adjacent Cyclic Ethers. Organic Letters 2001, 3 (13), 1989-1991. 10.1021/ol0159625
[12] Y. E. Hyun, H.-R. Kim, Y. Choi, L. S. Jeong, Stereoselective Synthesis of Anti-Hepatitis B Drug, Entecavir, through Regio- and Stereoselective Epoxide Cleavage Asian J. Org. Chem. 2017, 6, 1213. 10.1002/ajoc.201700032
No comments to display
No comments to display