First Total Synthesis of Granatumine A (T. R. Newhouse, 2019)
In this total synthesis the research group of Timothy Newhouse from Yale University (USA) describes not only the first synthetic route to granatumine A (1), it is also revealed that the structure of to closely related alkaloids have to be reassigned. Their synthesis was published in the Journal of American Chemical Society in 2019 and features a late stage pyran to pyridine interconversion as shown in Figure 1. The necessary key fragment 2 should be synthesized by Knoevenagel aldol condensation followed by spontanous cyclization of the two fragments 3 and 4.
 Figure 1: Retrosynthetic analysis for the total synthesis of granatumine A (1).
Figure 1: Retrosynthetic analysis for the total synthesis of granatumine A (1).
Synthesis of Coupling Fragments 3 and 4
For the synthesis of fragment 3 the Newhouse group opted for a six step sequence from racemic ionone 5 (see Scheme 1). Kinetic resolution through Jacobsen olefination yielded in 6 with moderate enantiomeric excess. 1,4 reduction to 7 was followed by ozonlytic oxidative conditions gave the free acid as intermediate, which opened the epoxide to lactone 8 in 51% yield after further oxidation. Then α,β-dehydrogenation with 92% yield was followed by epoxidation to 9. Finally epoxide opening led to fragment 3 in 90% yield.
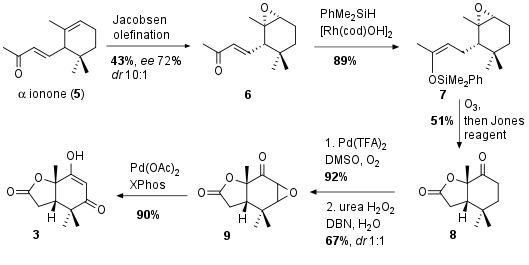 Scheme 1: Synthesis of dicarbonyl 3 in six steps from ionone 5.
Scheme 1: Synthesis of dicarbonyl 3 in six steps from ionone 5.
For fragment 4 symmetrical ketone 10 was transformed into the unsaturated ketone followed by aldol reaction wit furanaldehyde 11 yielding in racemic compound 12 (see Scheme 2). The authors again choose a kinetic resolution with the asymmetric base 13 to get the chiral compound 14 in 41% yield and 82% enantiomeric excess. The other enantiomer could successfully be transformed back by retro aldol reaction (not shown). With ketone 14 in hand the acetate was enolized and aldol reaction with the ketone followed by dehydrogenation led to pyrangolensolide (15). Note that this also describes the first total synthesis of this natural product conforming the absolute configuration. Finally allylic oxidation to azadaralide (16) and oxidation to 4 led to the desired fragment in six steps.
 Scheme 2: Synthesis of fragment 4.
Scheme 2: Synthesis of fragment 4.
Completion of Total Synthesis
As shown in Scheme 3 the both fragments were coupled to intermediate 17 which spontanously cyclize to pyran 2 as single isomer. Through DFT studies the authors argued that this regioisomer is thermodynamic preferred product. Then Luche reduction delivered alcohol 18 in 61% yield as single diastereomer. Together with hydroxylamine the pyran was transformed into pyridine 35% yield together with the side product 20. Note that 19 itself shows the proposed structure of xylogranatin F, however it turned out that the analytical data didn't fit to the data of the natural product. The authors could prove that there is a misassignment of the absolute configuration at the alcohol position (C3 epimer) for xylogranatin F and G (not shown). Furthermore, for the synthesis of granatumine A (1) the configuration had to be inverted. After several attempts, the transformation to the chloride and attack of NaOMe delivered 1 however as 1:1 diastereomeric but separable mixture at this position.
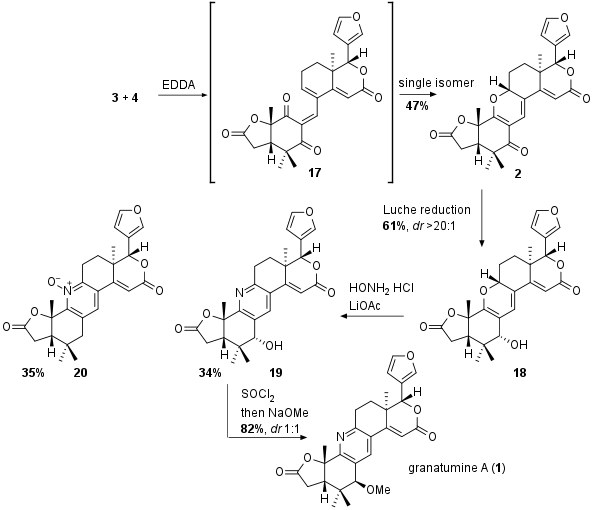 Scheme 3: Completion of the total synthesis of granatumine A (1).
Scheme 3: Completion of the total synthesis of granatumine A (1).
Published in A. W. Schuppe, Y. Zhao, Y. Liu, T. R. Newhouse J. Am. Chem. Soc. 2019, 141, 9191-9196. doi: 10.1021/jacs.9b04508