Synthesis of Cortistatin A from Prednisone (P. Baran, 2008)
For the enantioselective synthesis of complex alkaloid cortistatin A (1), the research group of Prof. Phil Baran at the Scripps Institute (USA) decided to start with prednisone (4) as shown in Figure 1. In their publication in the Journal of American Chemical Society in 2008 they argue that this steroid already posses 70% of the carbon atoms of the final product and is commercially available for a low price. However, for the transformation to cortistatin A a challenging ring expansion of derivative 3 is necessary to achieve the seven-membered ring of the alkaloid. Precursor 2 should then be functionalized to cortistatin A (1).
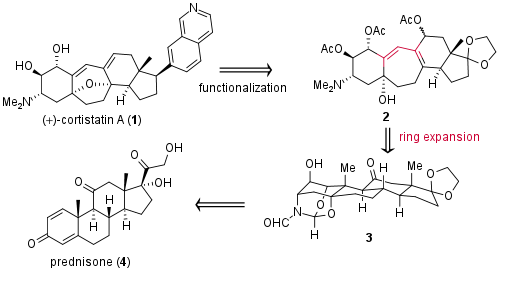
Figure 1: Retrosynthetic analysis of the synthesis of cortistatin A (1) from prednisone (4).
Synthesis of Key Fragment 3
To transform prednisone (4) into cortistatine A (1) the Baran group started with the regioselective reduction of ketone at the side chain with subsequent periodate cleavage, which itself led to another ketone which was immediately protected as ketale 5 in 92% yield over two steps. Regio- and diastereoselective epoxidation of the unsaturated ketone was followed by amination and generation of the formamide 6. After unsuccessfully using different acidic and basic methods for epoxide opening they reported a t-butylammonium acetate mediated regioselective epoxide opening. Finally, Mukaiyama hydration followed by protection of the newly generated alcohol together with the other free alcohol yielded in 3 after acetate cleavage with K2CO3 in 63% yield.

Scheme 1: Synthesis of ring-expansion precursor 3.
Ring Expansion of 3
For ring-expansion, as shown in Scheme 2 precursor was treated with PIDA and Br2 for in situ generation of AcOBr, which led to the selective dibromation of the methyl group. The free alcohol was afterward immediately protected to 8 to prevent ether formation. Enolate formation was then accompanied by cyclopropane formation due to the attack of the enolate at the dibromate for the synthesis of 9. Now the ring expansion took place by radical cyclopropane ring-opening with SmI2 and trapping of the enolate with TBCHD as Br+ source to get seven-membered ring 10. Lithium carbonate then led to the elimination and formation of 11 in 63% yield over two steps. Finally, reduction of the ketone and liberation of the dimethylamine with AlH3, cleavage of the TMS group with K2CO3 and acetate protection of the secondary alcohols under Steglich conditions yielded in 2 in 89% yield

Scheme 2: Ring expansion and functionalization to fragment 2.
Completion of the Synthesis
To complete the total synthesis the activation of acetate with MgBr2 was accompanied by the diastereoselective attack and elimination to the desired bicyclic ether. Deprotection of the ketone and the alcohols delivered cortistatinone (12) in 82% yield. In the final steps, the introduction of the isoquinoline moiety was enabled by the two-step formation of the vinyl iodide followed by Stille coupling to 13 in 53% yield. Reduction with Raney Nickel then led to the final natural product 1 in 50% yield (100% brsm).

Scheme 3: Completion of the synthesis of cortistatin A (1).
In conclusion, cortistatin A (1) was synthesized from commercially available prednisone (4) in around 3% yield using a challenging ring-expansion strategy. The authors underline the high importance of several transformations like the alcohol directed dibromination of the methyl group and the cleavage of the cyclopropane ring.
Published in R. A. Shenvi, C. A. Guerrero, J. Shi, C.-C. Li, P. S. Baran Journal of American Chemical Society 2008, 130, 7241-7243. doi: 10.1021/ja8023466
No Comments