Total Synthesis of Thebainone A (P. Metz, 2020)
Despite several total syntheses of opiates since the 50s, the construction of these complex alkaloids still challenges synthetic chemists nowadays. Recently the research group from Peter Metz from the TU Dresden successfully described a total synthesis of racemic thebainone A (1) in Organic Letters. Previously thebainone A could be synthesized by Gates during the first total synthesis of morphine and also by Michael Kerr and Marcus Tius in 1992 in 24 steps.
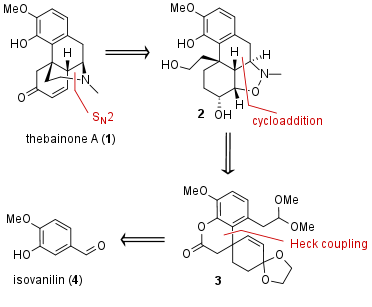
Figure 1: Retrosynthetic analysis of thebainone A (1).
From a retrosynthetic viewpoint amine-containing ring of thebainone A (1) should be closed by nucleophilic substitution as shown in Figure 1. The precursor should be derived from 2, which itself can be synthesized by cycloaddition. Finally, 3 is available from isovanillin (4).
Synthesis of Cycloaddition Precursor
At the beginning of the synthesis iodination of 4 delivered aldehyde 5 in 79% yield (see Scheme 1). Then homologation under Wittig conditions and acetalization led to 6. The phenolic compound then underwent esterification with literature known fragment 7 in excellent yield to achieve ester 8. The authors then describe a Heck cyclization leading to fragment 3 in 83% yield. Note that the double bond shifted due to the beta hydride elimination of the palladium intermediate (not shown) to the only possible direction.

Scheme 1: Synthesis of cycloaddition precursor 3.
[3+2] Cycloaddition
Under unusual Lewis acidic conditions, the carbonyl groups of 3 were deprotected and the aldehyde of 9 was then directly transformed into the nitrone, which underwent [3+2] cycloaddition to 10 in good diastereoselectivity as shown in Scheme 2. Due to insufficient stability, the compound was directly reduced to triol 2 in 56% yield from 3. The next four steps then were necessary to adjust the protecting groups for the completion of the total synthesis. All of them delivered excellent yields.

Scheme 2: Cycloaddition and further derivatization of the alkaloid core structure.
Completion of Total Synthesis
Reduction of the isoxazolidine using H2/Raney Ni delivered the secondary amine of 11 which in situ led to a nucleophilic substitution of the tosylate (see Scheme 3). This was followed by saponification to 12 in 92% yield over 2 steps. Direct conversion of 2 to 12 with the unprotected alcohols under Mitsonobu conditions gave not reproducible results and low yield. Reductive elimination of the diol yielded in alkene 13 in 81% yield over 2 steps. The amine was then protected by conversion into the carbamate 14. The following allylic oxidation gave poor yields due to poor regioselectivity and the alcohol was directly oxidized by Dess Martin periodinane to unsaturated ketone 15. A direct transformation to thebainone A (1) by reduction/oxidation was unsuccessful and therefore the ketone was protected as ketale. Then LiAlH4 reduction delivered 16 in 94% yield followed by acidic deprotection to the final natural product 1.
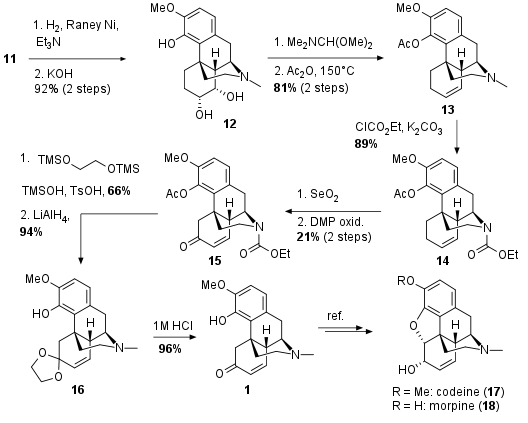
Scheme 3: Completion of the total synthesis of thebainone A (1).
In the end, Metz and coworkers could successfully synthesize thebainone A (1) in 22 steps. Their total synthesis features an intramolecular Heck addition and [3+2] nitroso alkene cycloaddition. Due to the fact, that 1 was already used as an intermediate for the total synthesis of codeine (17) and morphine (18) also a formal synthesis of these alkaloids was described.
Published in Y. Wang, A. Hennig, T. Küttler, C. Hahn, A. Jäger, P. Metz Organic Letters 2020, 22, 3145. doi: 10.1021/acs.orglett.0c00905
No comments to display
No comments to display