Total Synthesis of Mycinolide IV and Aldgamycin N (A. Fürstner, 2021)
Mycinolide IV (1, Figure 1) and aldgamycin N (2) are complex, bioactive polyketides. The macrolide core together with its glycosylation pattern was always a challenging task for synthetic chemists. The group of Alois Fürstner (MPI für Kohlenforschung, Germany) now published the total synthesis of both polyketides in two articles in the journal Angewandte Chemie. The overall goal of their synthetic study was to describe a unified approach to both polyketides, which should also enable the synthesis of closely related structures (not shown). As shown in Figure 1, the macrolides should be available by the coupling of two major fragments 5 and 6. The precursors 3 and 4 for mycinolide IV and aldgamycin N, therefore, can be synthesized using the same methods, with slight modifications in the synthetic route.

Figure 1. Retrosynthetic analysis for the total synthesis of mycinolide IV (1) and aldgamycin N (2).
Synthesis of fragments 5 and 6
The eastern part of the macrolide was constructed as shown in Scheme 1. Starting from allylic alcohol 7, Sharpless epoxidation and ring-opening gave 8 in acceptable enantioselectivity, however, with a small yield. Then TBDPS protection of the primary alcohol was followed by TES protection of the remaining free alcohol. Finally, reductive iodination gave 5 in good yields. For the synthesis of mycinolide IV (1) iodoalkene 5b was coupled with TMS-alkyne yielding in 9 after deprotection. For the synthesis of aldgamycin N (2), fragment 10 was prepared in a similar way.
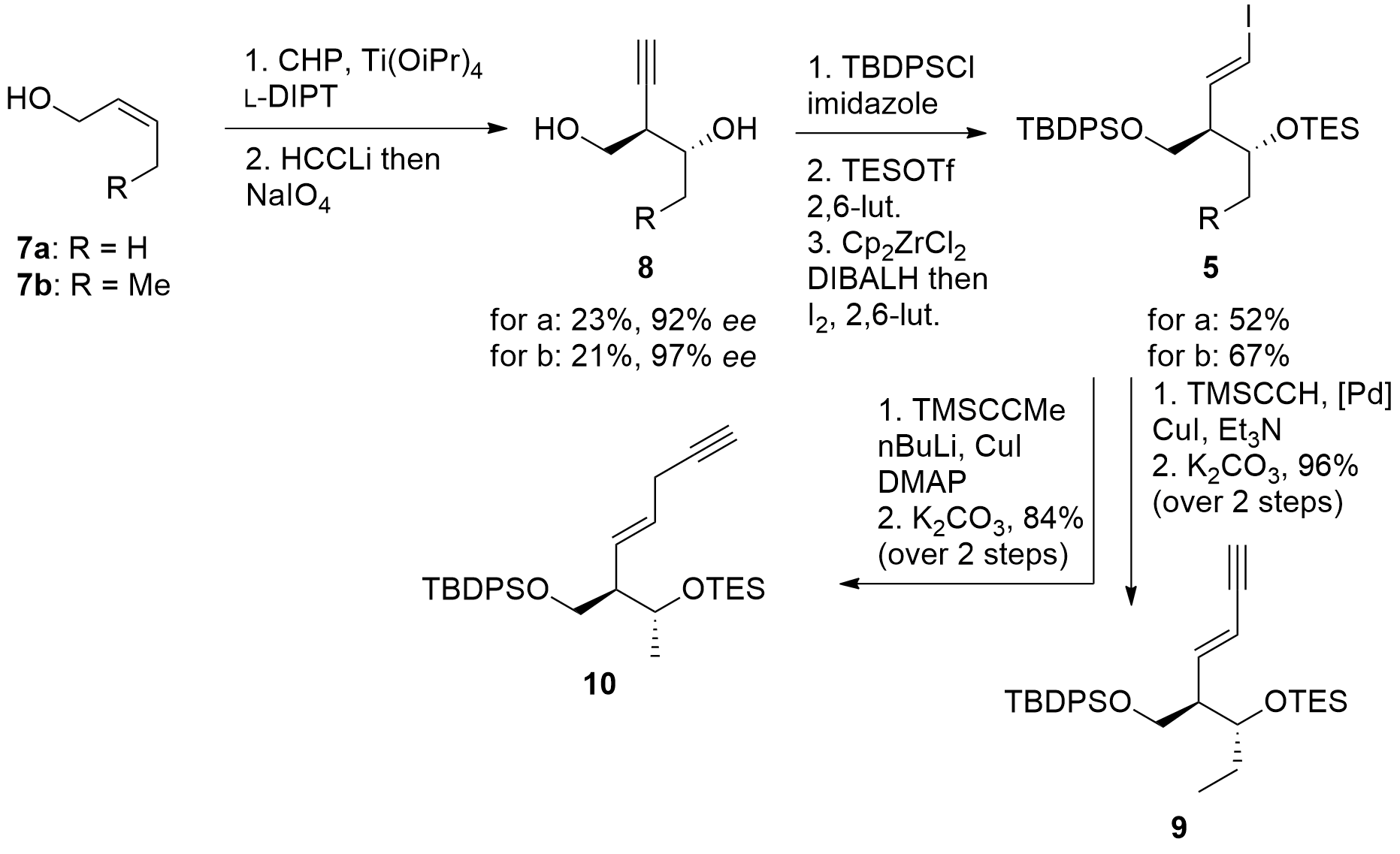
Scheme 1. Synthesis of the eastern fragment 5.
The western part of the macrolide could be constructed in a straightforward way from known compound 11 as shown in Scheme 2. Reduction gave the racemic alcohol, which was transformed into entantio-enriched acetate 12 by enzymatic resolution. Deprotection under unusual conditions (MeLi) and oxidation gave aldehyde 13. For the vinylogous Mukaiyama aldol reaction, the shown oxazaborolidine could successfully catalyze the reaction and gave 6 in usable dr.

Scheme 2. Synthesis of western fragment 6.
Total synthesis of mycinolide IV (1)
With both fragments in hand, the Fürstner group now was able to finish the synthesis of mycinolide IV in a few steps as shown in Scheme 3. First, MOM protection of the free alcohol of 6 was followed by hydroformylation which yielded 14. Then, western fragment 9 was introduced by the attack of the lithiated species. TES deprotection then gave diol 15. For ring-closing, the authors reported that they were not able to cleave the methyl ester. Therefore, a transesterification approach was needed using a stannoxane complex yielding in fragment 3 albeit with a low yield. [Ru] catalyzed redox isomerization and acidic deprotection then finally yielded in mycinolide IV (1).
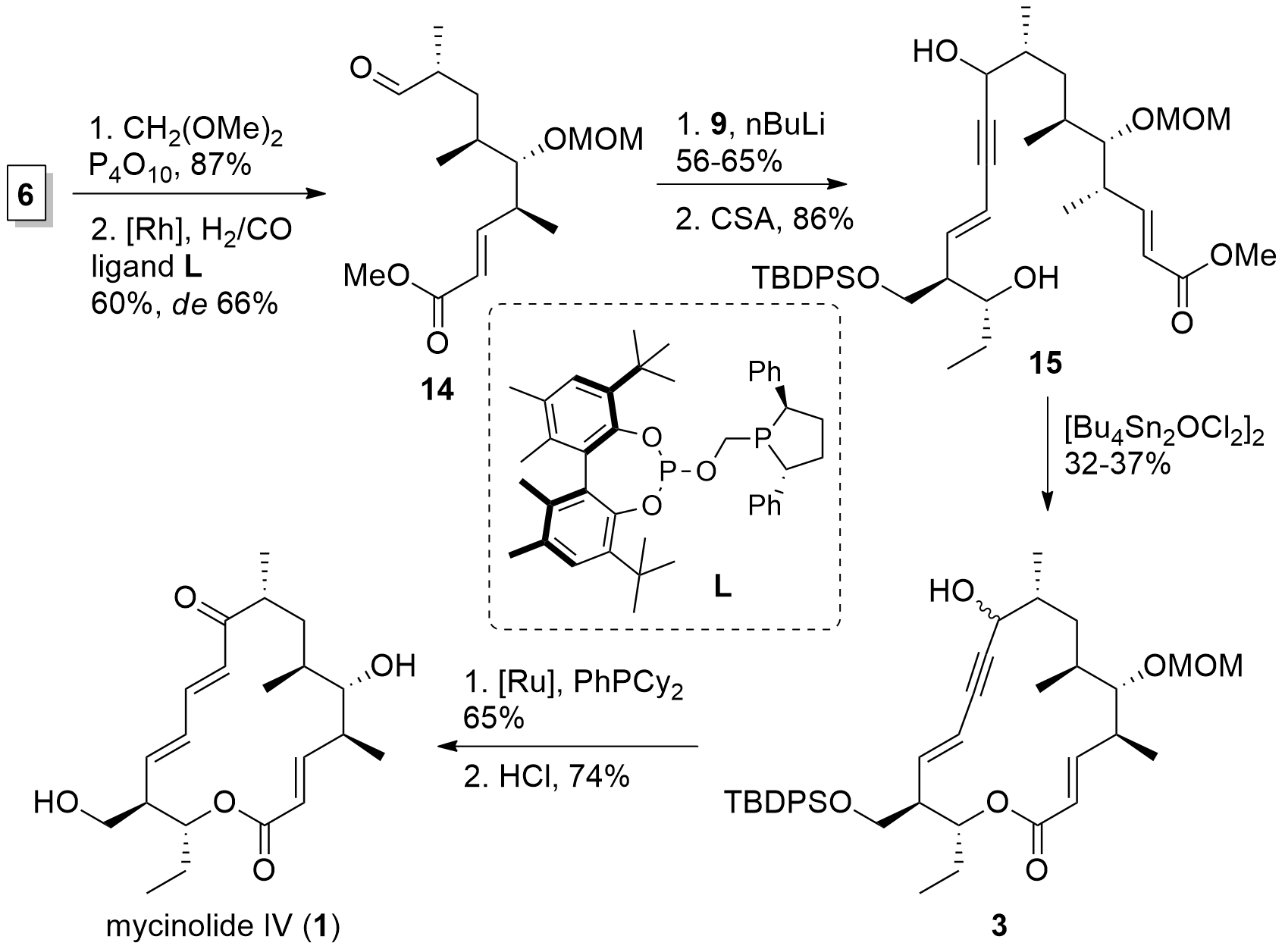
Scheme 3. Total synthesis of mycinolide IV (1).
Total synthesis of aldgamycin N (2)
For aldgamycin N (2) a much more complex approach was needed by Fürstner and co-workers. One major challenge was the synthesis of both sugar moieties. First, aldgarose precursor 20 was targeted as shown in Scheme 4. Therefore, fragment 16 was transformed into the silyl enol ether and the following hetero Diels-Alder reaction with subsequent acidic deprotection gave unsaturated ketone in good enantioselectivity and yield. Diastereoselective epoxidation under basic conditions was followed by TIPS protection and the addition of vinyl magnesium bromide yielding 18 in 46% yield over 3 steps. The terminal alkene was then epoxidized and the epoxide was opened by hydride. The diol was then transformed into the carbonate, and at the same time, TIPS protection group was cleaved. The free alcohol was again protected, this time as acetate and also the acetal was acetate protected yielding in 19 in 5 steps from 18. Then the acetal was further changed to the shown trichloroacetimidate as a suitable coupling partner.
The mycinose sugar 25 was synthesized from 21 by first bromination and then methylation to fragment 22. Then alkene reduction was followed by debromination. Reduction of the ester to the aldehyde was followed by the formation of the desired pyranose form 21. Again, transformation into the trichloroacetimidate 25 then facilitated the following coupling.
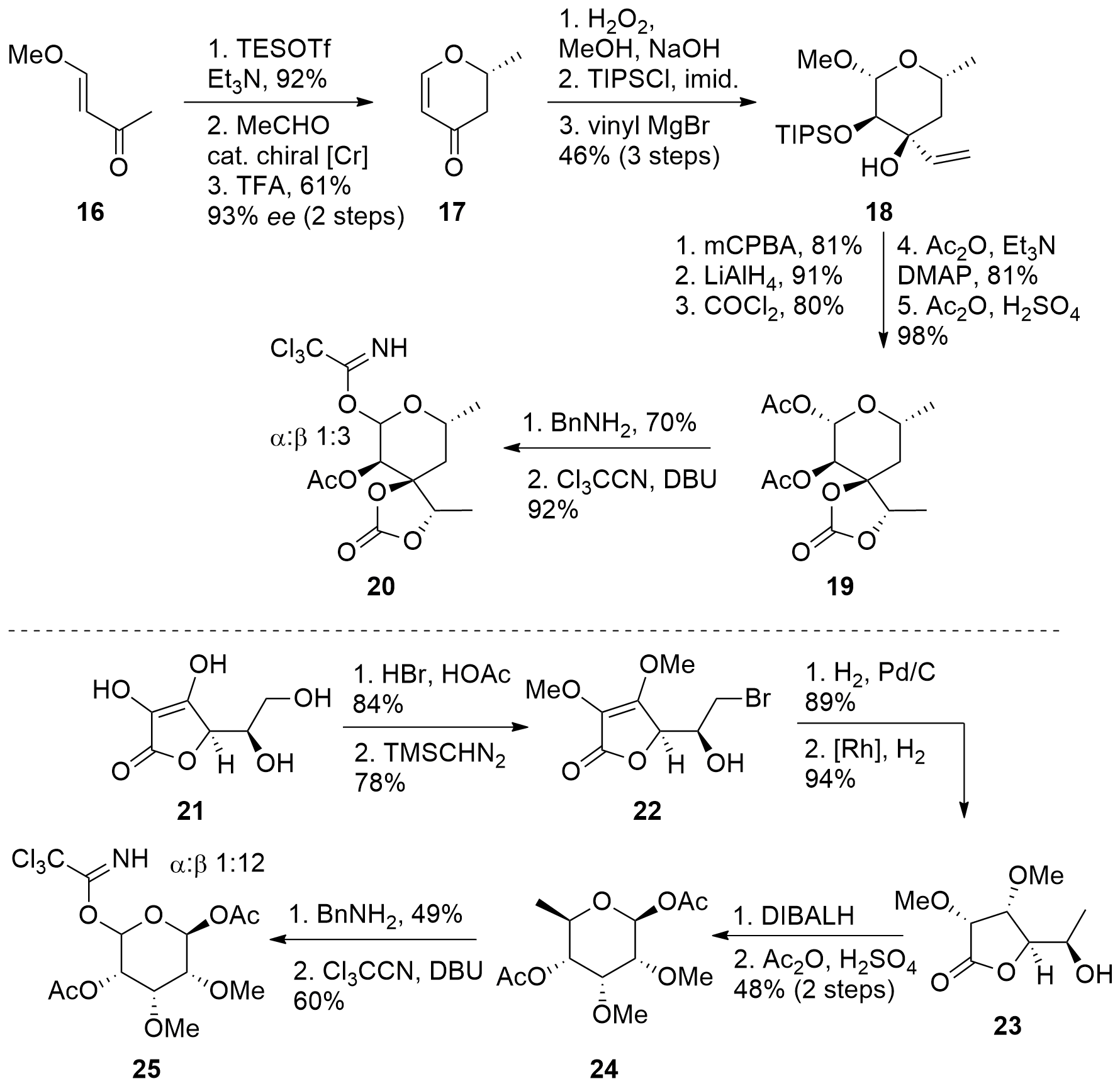
Scheme 4. Synthesis of both sugars for the total synthesis of aldgamycin N (2)
Finally, with all fragments in hand, the Fürstner group was able to assemble aldgamycin as shown in Scheme 5. First western fragment 6 was transformed into ketone 26 by PMB protection and oxidation of the alkene. The addition of the eastern fragment was achieved using again the lithiated fragment 10 followed by acidic deprotection. Both diastereomers could be separated in this step and both were used for the determination of the absolute configuration of the tertiary alcohol (only the correct one is shown). Again, stannoxane-mediated transesterification successfully enabled the macrolide formation. With 4 in hand, the first sugar moiety was introduced. Therefore tertiary alcohol was TES-protected and PMB was deprotected. The addition of trichloroacetimidate under Lewis acidic conditions indeed gave the desired aldgarose 29 as a single isomer after silyl deprotection. The authors highlighted that a late-stage introduction of the sugar was not possible due to the attack of free alcohol to the ketone (not shown). Alkyne 29 was then transformed into an unsaturated ketone by first hydrostannylation followed by oxidation to the ketone 30. For the completion of the total synthesis, sugar 25 was coupled with the free, primary alcohol, however, only low stereocontrol was observed. The desired isomer could be isolated and carbonate cleavage liberated aldgamycin N (1).

Scheme 5. Completion of the total synthesis of aldgamycin N (2).
In total, the Fürstner group successfully could establish a synthetic route to a few macrolides. They could prove the suitability of this route with the synthesis of mycinolide IV (1) and aldgamycin N (2). Both polyketides were synthesized using a unified approach. During the synthesis, the authors describe several challenges like the asymmetric vinylogous aldol reaction, the transesterification and the synthesis of aldgarose.
Published in: B. Herlé, G. Späth, L. Schreyer, A. Fürstner Angew. Chem. Int. Ed. 2021 doi: 10.1002/anie.202016475 and G. Späth, A. Fürstner Angew. Chem. Int. Ed. 2021 doi: 10.1002/anie.202016477
For another total synthesis by Fürstner et al see: First Total Synthesis and Structural Revision of Prorocentin (A. Fürstner, 2023)
No comments to display
No comments to display