Total Synthesis of Alterbrassicicene C (J. L. Wood, 2022)
Alterbrassicicene C (1) is a diterpenoid with an unusual tetracyclic core structure. John L. Wood (Baylor University, US) and co-workers now reported the first enantioselective total synthesis of this natural product in the Journal of American Chemical Society (10.1021/jacs.2c12275). As shown in Figure 1, their retrosynthetic analysis showing key intermediate 2 which should be available by a reaction cascade from 3 in a single step. Ether 3 itself should be prepared from 4 by etherification and 1,2-migration. Finally, cyclopentanes 5 and 6 should deliver 4 by nucleophilic addition and ring-closing metathesis (RCM).

Figure 1: Retrosynthetic analysis for alterbrassicicene C (1) by J. L. Wood et al.
Synthesis of Building Blocks and Coupling
Both building blocks 5 and 6 were synthesized in a straightforward fashion from literature described compounds as shown in Figure 1. In detail, ketone 7 could be functionalized in alpha position after enolate formation. Then the terminal double bond was isomerized using Grubbs-II catalyst and oxidative C-C bond cleavage delievered aldehyde 8 in 3 steps. Formation of the enolate was then followed by triflation and Stille coupling to furnish the first building block 5.
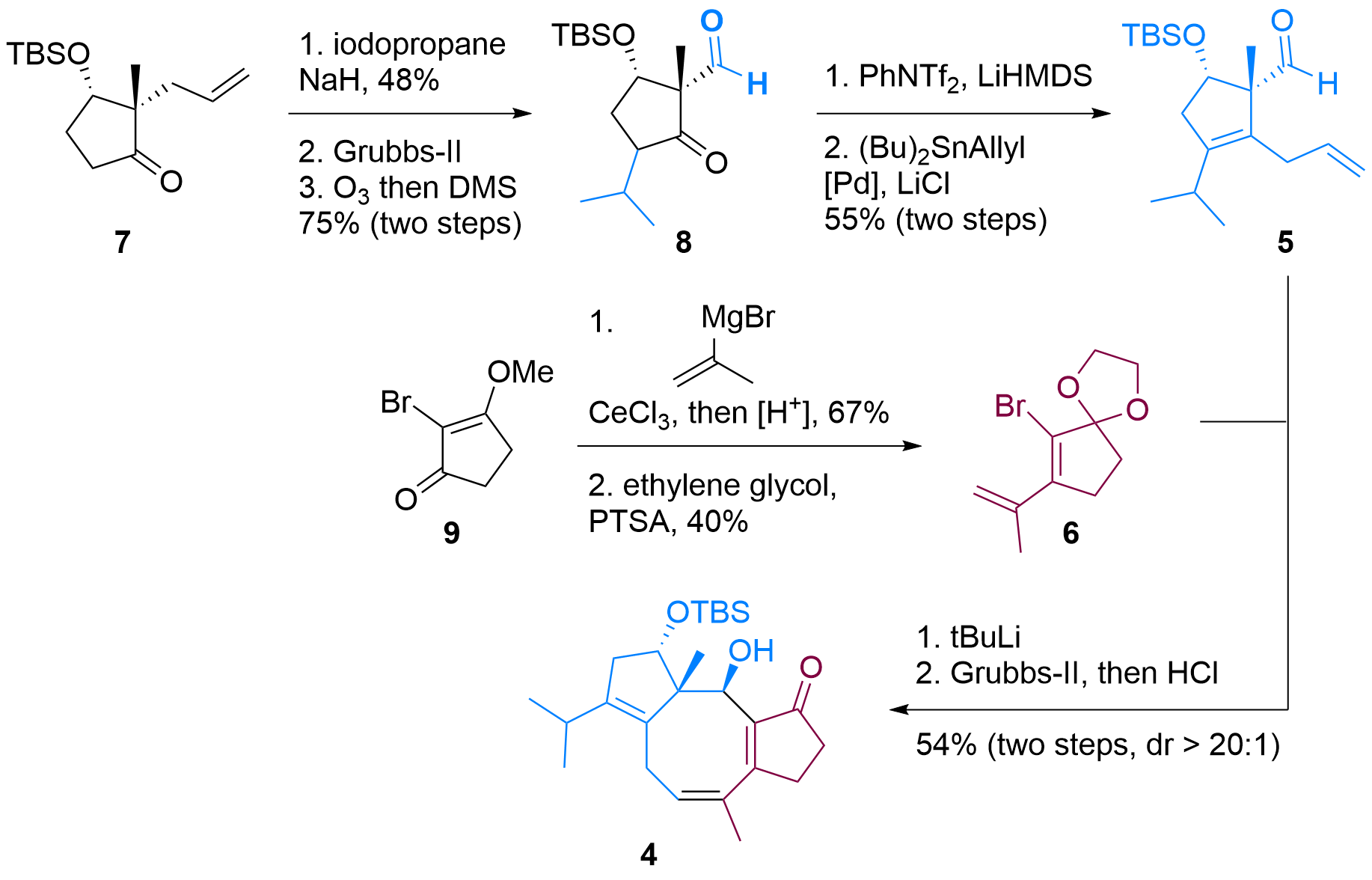
Figure 1: Synthesis of building blocks 5 and 6 and coupling to 4.
Bromide 6 was synthesized in two steps from 9 by first Grignard addition, then deprotection and condensation. Finally, the ketone was masked as ketal. After treating with tBuLi, lithiated 6 added to 5 in high diastereoselectivity and acidic deprotection revealed tricyclus 4.
Etherification and 1,2-Migration
As shown in Scheme 2, diene 4 was treated with NBS which initiated the etherification to 10 with silyl ether cleavage in the same step. The following 1,2-migration initiated by Ag(TFA) was high yielding, however, the authors noted that the attack of alcohol and the formation of the bis-ether 11 was not intentionally. Therefore, another steps was necessary to open the tetrahydrofurane to achieve 12 under selective chlorination. Now, epoxidation and oxidation yielding in key intermediate 3.

Scheme 2: Synthesis of key intermediate 3.
Oxa-Michael Cascade and Late-Stage Functionalization
With highly functionalized bridged tricyclus 3 in hand, the key reaction using DBU was probed. As shown in Scheme 3, first enolate formation and retro-oxa-Michael reaction furnished intermediate 13. Then oxa-Micheal reaction to delivered 14 which then finally eliminates to 2. After successful formation of the core structure, epoxidation was followed by the formation of the enol triflate 15 using standard conditions. Protection of the alcohol was necessary for the subsequent Stille coupling. Finally, epoxide opening was followed by a 1,2 hydride shift and double bond isomerism. Then deprotection radical dechlorination delievered alterbrassicicene (1).

Scheme 3: Oxa Michael cascade and finalization of the total synthesis of 1.
In this remarkable total synthesis of the diterpenoid (1) by John L. Wood and co-workers, the product could be synthesized in 20 steps (longest linear sequence from literature known compounds).
Published in: Noah J. Sims, Weston C. Bonnet, Danielle M. Lawson, John L. Wood Journal of American Chemical Society 2022 (10.1021/jacs.2c12275)
For another synthesis by John L. Wood, see the total synthesis of phyllantidine in 2020.
No comments to display
No comments to display