First Total Synthesis of Daphnezomines A and B (C. Li, 2020)
Alkaloids from the plant genus Daphniphyllum are known for their complex structure and attracted many synthetic chemists for several decades. Now, Chao Li (李超) and co-workers from the National Institute of Biological Science in Beijing (China) were able to report the first total synthesis of daphnezomine A (1) and B (2, see Figure 1) in the journal JACS. From a retrosynthetic point of view, the Li group planned a late-stage tautomerization to yield the desired aza-adamantane core. The precursor 3 should be available from 4 by a challenging radical cyclization. Finally, 4 could be disconnected to fragment 5 which itself is available from carvone.

Figure 1: Retrosynthetic analysis for the total synthesis of daphnezomine A (1) and B (2).
Synthesis of Cyclization Precursor
Inspired by a literature-known sequence, the Li group decided to use (S)-carvone (6, Scheme 1) as their chiral precursor. The reduction of the two double bonds was followed by the formation of the silyl enol ether. Then allylic Sharpless amination allowed the introduction of the desired amine in 41% yield which was then transformed into the tertiary amine 7 using allyl bromide. Palladium-catalyzed oxidative cyclization allowed the synthesis of a bicyclic system. Then the ketone moiety has to be protected for the following hydroboration to get fragment 8. The hydroboration then was directly followed by a Suzuki-Miyaura coupling with iodide 9 and deprotection of the ketone to 10. Note, that although the hydroboration yielded in the desired diastereomer at C6, the deprotection of the ketone moiety led to complete epimerization at C11.
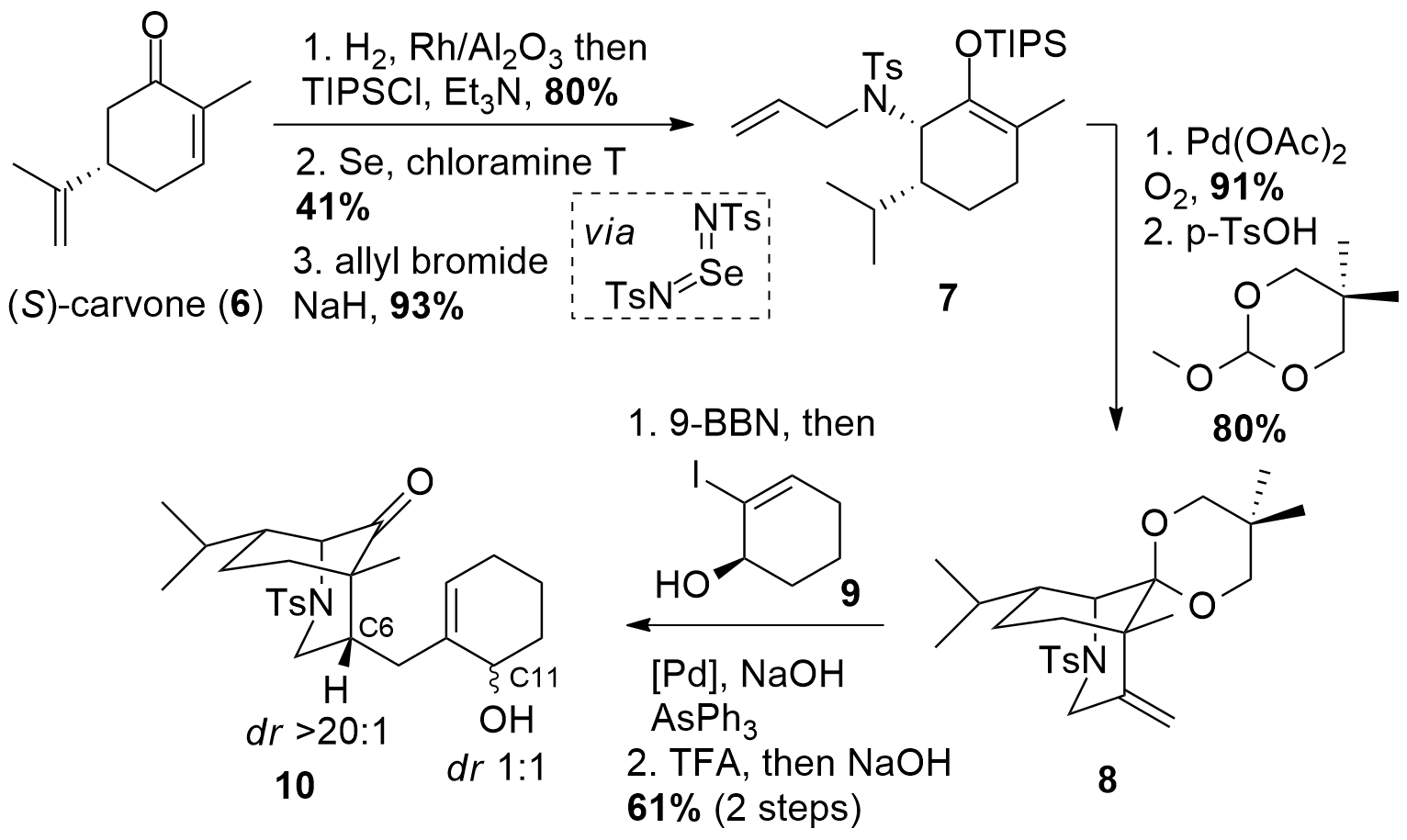
Scheme 1: Synthesis of intermediate 10 from carvone (6).
With fragment 10 in hand, the Li group further functionalized the ketone by organometallic addition in 80% yield as shown in Scheme 2. Then deprotection of the amine and Boc protection was followed by alcohol oxidation to unsaturated ketone 11 in 79% yield. Both diastereomers could be transformed into 4 under dehydration conditions using Burgess reagent in very high yield. An unusual benzyl deprotection with in situ oxidation to the acid was followed by methyl ester formation and Boc deprotection to get cyclization precursor 12. 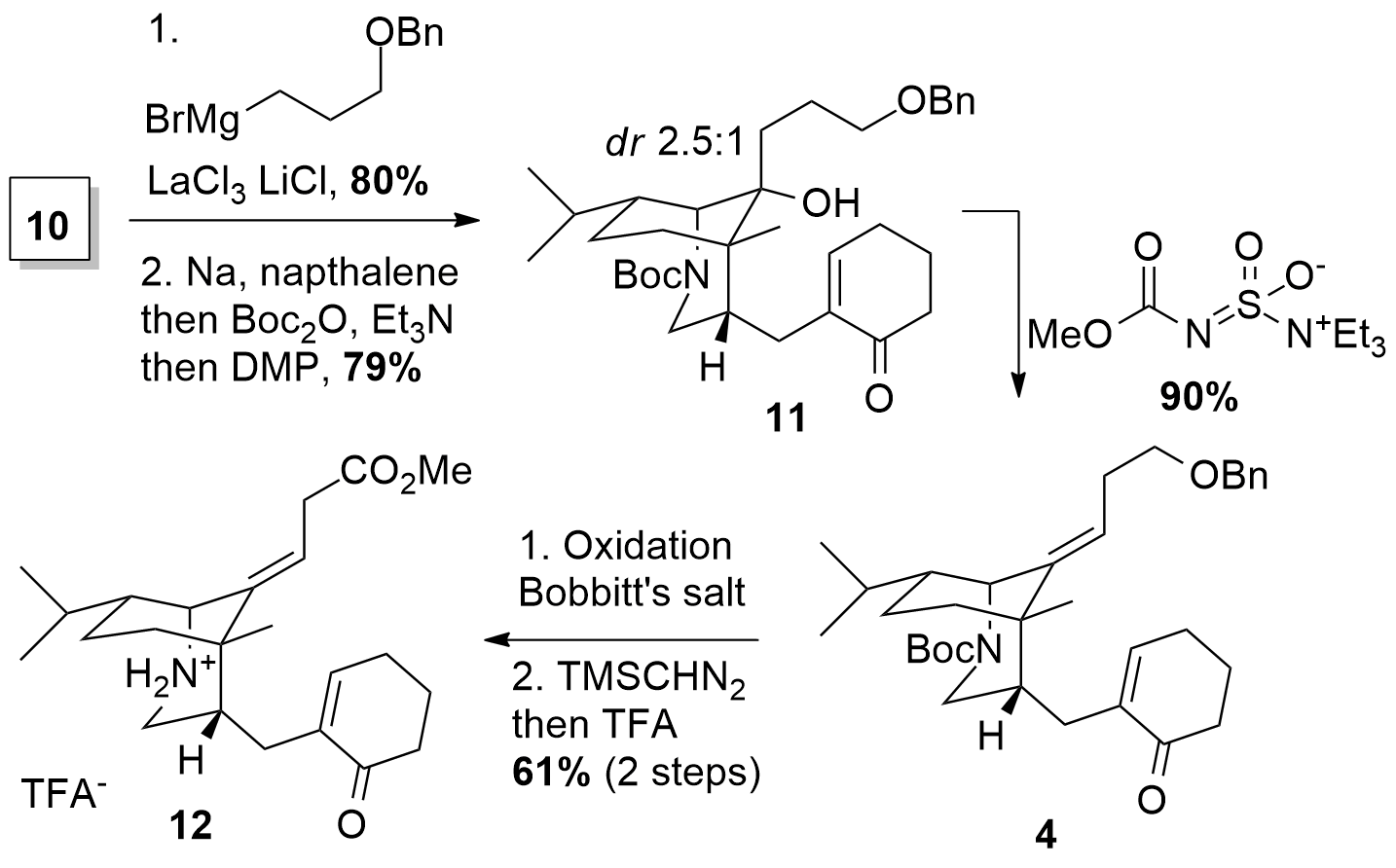
Scheme 2: Synthesis of cyclization precursor 12.
Radical Cyclization and Completion of Total Synthesis
For the following 6-endo-trig cyclization the Li group followed three different strategies for cyclization (Lewis acidic ene cyclization, anionic cyclization & radical cyclization using HAT). In the end, Hydrogen Atom Transfer (HAT) promoted radical cyclization by conjugate addition led to the desired C-C bond formation in 31% using reductive conditions. This delivered the free amine 3, the (C10 epi) tautomer of the desired natural product. Consequently using acidic conditions not only led to C10 epimerization but also yielded in daphnezomine A (1) in 53% yield. If methanol was used as solvent a mixture of 1 and 2 was achieved, however, 1 could easily be transferred into 2 by methyl ester formation.
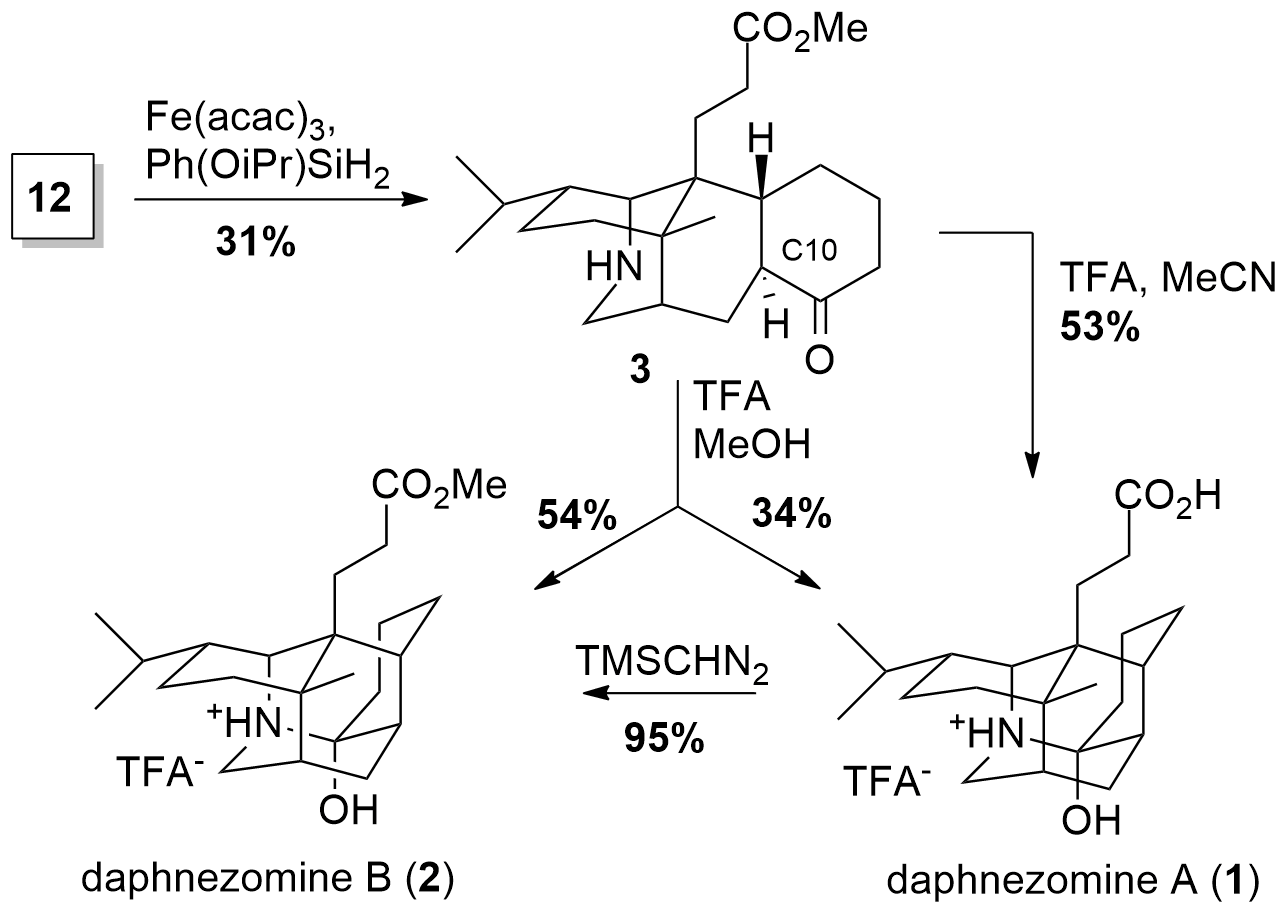
Scheme 3: Completion of the total synthesis of daphnezomine A (1) and B (2).
In Conclusion, the Li group successfully described the total synthesis of daphnezomine A (1) and B (2) using an adventurous late-stage cyclization and a challenging Suzuki Miyaura sp2-sp3 coupling. In total 14 steps were needed to synthesize the alkaloid from (S)-carvone in a diastereoselective manner.
Published in: G. Xu, J. Wu, L. Li, Y. Lu, C. Li Journal of the American Chemical Society 2020, 142, 15240-15245. doi: 10.1021/jacs.0c06717
No Comments