Total Synthesis of Ajudazol A (D. Menche, 2020)
Ajudazol A (1) is a secondary metabolite of mycxobacterial origin which containing an isochromanone ring system, an oxazole, and a polyene moiety. After the total synthesis of ajudazol B in 2012, the research group of Prof. Dirk Menche (University Bonn, Germany) is now able to report the synthesis of a second member of this natural product family in the journal Organic Letters. From a retrosynthetic point of view, the Menche group decided to use three main fragments, which should be assembled in a late-stage synthesis to the final natural product by cross-coupling. Fragment 4 is already known and isochromanone fragment 2 could be achieved in two steps from an intermediate which was used in the previous synthesis. However, the connection of 2 and 3 remained challenging similar to other sp3-sp2 cross-couplings in total synthesis.

Figure 1: Retrosynthetic analysis of the total synthesis of ajudazol A (1).
Synthesis of Key Fragments 2,3 and 4
As already mentioned the synthesis of 11 (see Scheme 1) was reported previously by Menche et al. for the synthesis of ajudazol B. As a general precursor acid 5 was allyl protected on the alcohol and acid moiety. However, the resulting ester was then directly cleaved and transformed into amide 6 in two steps. The following ortho lithiation proved to be regioselective and quenching with chiral Anderson reagent yielded in 8. Again, lithiation and addition of aldehyde 9 then finally gave 10 in 71% yield. The cleavage of sterically hindered amide proofed to be challenging and was done in a two-step procedure. TBS protection of free alcohol was followed by allyl deprotection and saponification. Under these conditions, TES ether was cleaved simultaneously leading directly to the isochromanone 11. For the synthesis of ajudazol A (2), the phenol was TBS protected followed by transformation of the terminal alkene into the iodide 2 by Lemieux Johnson oxidation, reduction, and Appel reaction.

Scheme 1: Synthesis of fragment 2.
For the synthesis of fragment 4, the same route as for the synthesis of ajudazol B was used as shown in Scheme 2. Alcohol 12 was oxidized to the aldehyde and Wittig reaction gave unsaturated ester 13 in 60% yield. Then reduction to the alcohol and Appel reaction yielded in bromide 14. At the same time, methyl acetoacetate 15 was enolized and methylated to 16 in excellent yield. This was followed by saponification and Steglich amidation for the synthesis of coupling partner 17. After deprotonation of the amide, alkylation gave 18 in 68% yield. Finally, rhodium-catalyzed hydroboration was necessary to achieve fragment 4.

Scheme 2: Synthesis of Eastern fragment 4.
Usually, oxazoles are prepared through cyclodehydration during total synthesis. For a more convergent synthesis, the Menche group decided to use a readily commercial available oxazole 19 as their central fragment (see Scheme 3). Whereas the first lithiation of the heterocycle at H-2 gave access to the alkylation product 20, the second lithiation happened at the undesired H-5 position as previously expected yielding in oxazole 21. The Menche solved this problem by using a so-called halogen-dance reaction in which H-4 was deprotonated by LDA. This intermediate then should rearrange to 3. The authors describe that catalytic amounts of 22 were necessary to prevent competing lithium iodide exchange.

Scheme 3: Synthesis of oxazole fragment 3.
Completion of the total synthesis
With all three fragments in hand, the Menche group first examined the challenging sp3-sp2 cross-coupling between fragments 2 and 3 as shown in Scheme 4. After an extensive screening of conditions on a model system, the coupling was successful using Negishi cross-coupling conditions. Deprotection and oxidation of TES ether 23 yielded in an aldehyde, which was directly transformed into 24 using a Wittig reaction. For the completion of the synthesis, Suzuki coupling with fragment 4 in 63% yield was followed by TBS deprotection to yield ajudazol A (1).
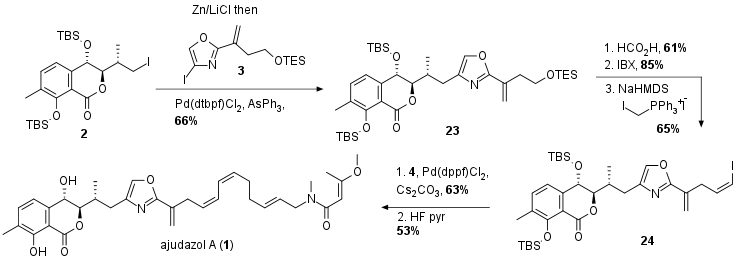
Scheme 4: Completion of the total synthesis of ajudazol A (1).
In summary, the Menche group successfully described the total synthesis of ajudazol A (1) in 17 steps (longest linear sequence). Furthermore, they also examined the synthesis of an analog (not shown) which retains the biological activity but is synthesized in a dramatically shorter route (9 steps).
Published in: P. Wollnitzke, S. Essig, J. P. Gölz, K. von Schwarzenberg, D. Menche Organic Letters 2020, 22, 6344-6348. doi: 10.1021/acs.orglett.0c02188
For other total synthesis by Menche et al see the total synthesis of archazolid F (2019) and tusceron D and E (2019).
No Comments