Total Synthesis of Archazolid F (D. Menche, 2019)
After the synthesis of archazolid A and B Menche et al. from the University Bonn published the synthesis of archazolid F (1), the most biological active member of the archazolid family. The retrosynthetic analyses shows that the authors decided to close polyketide macrocycle by ring-closing metathesis (see Figure 1). The precursor for the RCM should be synthesized from 3 different fragments. The northern fragment 2, the southern fragment 3 and commercially available acid 4.

Figure 1: Retrosynthetic analysis for the total synthesis of archazolid F (1).
Synthesis of Northern Fragment
As shown in Scheme 1 the northern fragment 2 was synthesized from aldehyde 5. After Wittig olefination to 6 the substrate was reduced to highly volatile aldehyde 7. At the same time lactate 8 was transformed into 10 by coupling with Weinreb amide and successive treatment of 9 with EtMgBr and coupling with Bz2O under basic conditions. Then 10 was coupled in an anti selective Paterson aldol reaction with 7 to get 11 which was instantly protected as TBS ether.

Scheme 1: Synthesis of the northern part of archazolid F.
Synthesis of Southern Fragment
The southern fragment was synthesized in 7 steps from commercially available bromide 23 (see Scheme 2). Literature known transformation into to Grignard reagent followed by coupling with isovaleraldehyde yielded in racemic alcohol 24. Reduction and oxidation deliviered the dicarbonyl 25. Brown crotylation set the desired stereocenter at C23 albeit with very low yield and enantiomeric excess. The free alcohol was then protected as TES ether. Ketone 27 was then subjected to a selective CBS reduction yielding in alcohol 28. Acetate protection to 29 was finally followed by cross metathesis with acroleine to the final southern fragment 3 in high yield.
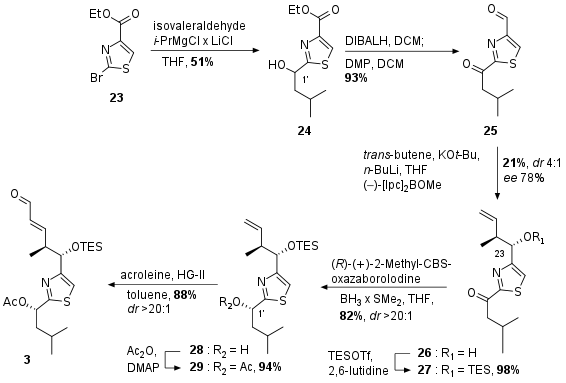
Scheme 2: Synthesis of the southern fragment 3.
Completion of Total Synthesis
With the final fragments in hand the authors described an unusual aldol condensation mediated by lithium diphenylamine as shown in Scheme 3. After aldol reaction between 2 and 3 condensation was triggered by transformation of the free alcohol to the acetate followed by DBU initiated elimination to unsaturated ketone 30. Diastereoselective reduction and methylation led to 31 in good yields. Deprotection in 1' position was followed by literature known carbamate formation in 88% yield. Then desilylation and simple DCC mediated coupling with butenoic acid yielded in RCM precursor 32. The ring closing was catalyzed by metathesis catalyst 33, a derivative of the Stewart Grubbs catalyst. The final natural product was isolated after desilylation and HPLC seperation in moderate yield.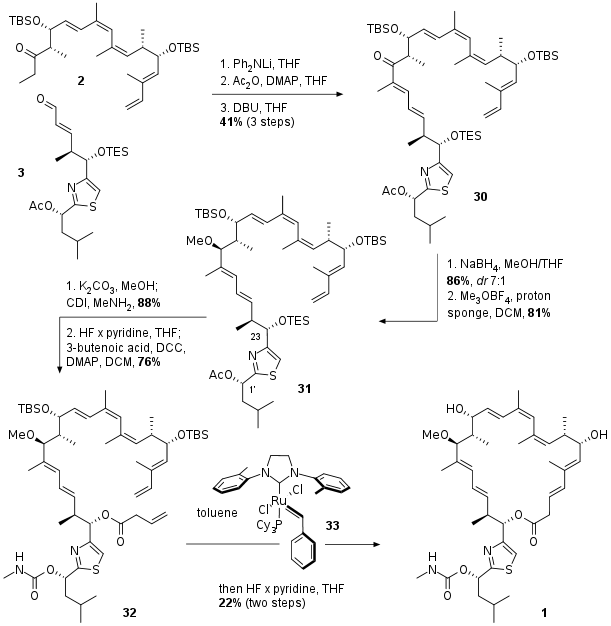
Scheme 3: Completion of the total synthesis of archazolid F (1).
Published in S. Scheeff, D. Menche Org. Lett. 2019, 21, 271-274. doi: 10.1021/acs.orglett.8b03715
No Comments