Total Synthesis of Bilobalide (R. Shenvi, 2019)
Bilobalide (1, see Figure 1) is a sesquiterpene which can be found in Gingko leaves and is related to the ginkgolide diterpenoids. Recently the research group of Ryan Shenvi from the Scripps Research Institute (USA) published in Nature a short total synthesis of this complex natural product. From a retrosynthetic point of view, they envisioned a late-stage oxidation of 2 to the final natural product. This would lead to a more simplified precursor with a symmetrical dilactone moiety, however, it was unknown whether a regio- and stereoselective oxidation would be possible. Lactone 2 itself should be prepared staightforwardly from 3, which itself is available from cyclopentene 4.

Figure 1: Retrosynthetic analysis of bilobalide (1).
Synthesis of Key Fragment 2
As shown in Scheme 1 cyclopentene 4 was assembled by radical cyclization of 11. In this 5-exo-trig cyclization, 4 could be synthesized in 60% yield. Precursor 11 itself was synthesized by a challenging enantioselective Reformatsky reaction using chiral BOX catalyst 10, which was developed for this transformation. Even with only acceptable diastereomeric control purification and recrystallization led to 4 as a single enantiomer. Bromide 7 was readily available from 5 by bromination and Wittig reaction with 6 which gave 7 in both good yields after conjugation of the double bond. Coupling partner 9 was synthesized from 8 using Vilsmeier-Haack conditions. Finally, Drago-Mukaiyama hydration under optimized conditions gave alcohol 12 in 67% with moderate diastereoselectivity.

Scheme 1: Assembling of cyclopentene 4 by radical cyclization of Reformatsky product 11.
In the next step, the Shenvi group had to protect the newly generated alcohol to prevent dehydration. They described an oxetane formation under acidic conditions which led to the stabilization of the tertiary alcohol as shown in Scheme 2. Using a chiral Binol derived phosphoric acid they could achieve good diastereoselectivity which was necessary for the following steps because of dehydration of the epimer of 13 (not shown). Secondary alcohol was then oxidized using IBX leading to a 1,3-dicarbonyl which undergo addition of alkyne using Waser's reagent. Reduction then gave alcohol 3 in 60% yield over 3 steps. The authors noted the highly challenging screening of suitable conditions for the diastereoselective synthesis of this highly complex bicyclus. Then the formation of the alkyne borate was followed by oxidation which directly led to the lactone 14. Reductive deprotection of the esters and acidic acetal cleavage led to key fragment 2 in 90% yield.
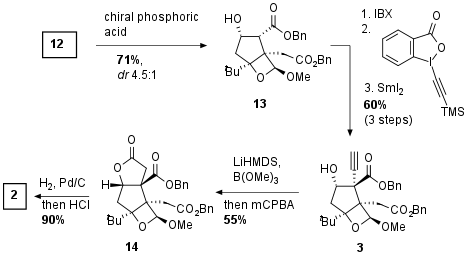
Scheme 2: Synthesis of key fragment 2.
Late-Stage Oxidation to Bilobalide
As already mentioned late-stage oxidation of 2 proofed to be challenging due to the inaccessibility of the position which had to be oxidized. The authors describe that various conditions led to the oxidation at the wrong position (regioselectivity) or to decomposition (see Scheme 3). Interestingly, they discovered that using KHMDS a translactonization occurred, however, oxidation still gave neo bilobalide 15 in quantitative yield. After an extensive search, they found that the protection of tertiary alcohol could successfully activate the desired lactone. Furthermore, the already described translactonization led to a ring-system like 16 where the cavity is unfolded and therefore oxidation at the desired position is actually possible. With these conditions in hand, late-stage oxidation could be successfully conducted liberating bilobalide 1 after acidic deprotection and re-translactonization.

Scheme 3: Late-stage oxidation of 2 at hindered position led to bilobalide (1).
In conclusion, the Shenvi research group could successfully describe a total synthesis of bilobalide (1) which is not only innovative but also inspiring for the community by solving problems that seemed to be unsolvable using conventional organic chemistry.
Published in M. A. Baker, R. M. Demoret, M. Ohtawa, R. A. Shenvi Nature 2019, 575, 643-646. doi: 10.1038/s41586-019-1690-5
No comments to display
No comments to display